
¿Qué es Hiperplasia Suprarrenal Congénita?
La Hiperplasia Suprarrenal Congénita (HSC), también conocida en la literatura clásica como “síndrome adrenogenital”, es una de las endocrinopatías más frecuentes en la infancia. Es un trastorno hereditario, de forma autonómica recesiva.
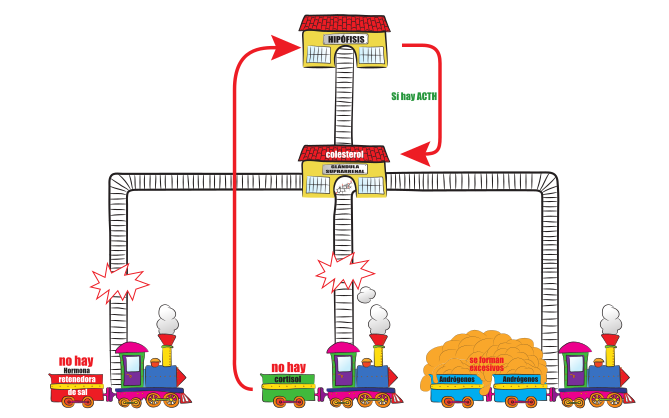
Las glándulas suprarrenales, localizadas en las parte superior de cada uno de los riñones, producen las hormonas cortisol, aldosterona y andrógenos que son esenciales para la vida.
Las personas con hiperplasia suprarrenal congénita tienen un déficit de alguna de las enzimas que regulan la síntesis de dichas hormonas, lo que dará lugar a un determinado déficit hormonal.
Todos los tipos de hiperplasia suprarrenal congénita tienen en común la ausencia parcial o total de cortisol, lo que provocará la elevación de ACTH y el crecimiento hiperplásico de las glándulas suprarrenales.
La hiperplasia de las glándulas suprarrenales se acompañará en ocasiones, del aumento en la producción hormonal (la que la glándula pueda producir a pesar del defecto enzimático).
La hiperplasia suprarrenal congénita puede afectar tanto a los niños como a las niñas. Alrededor de 1 de cada 10,000 a 18,000 niños/as en la forma clásica y 1 de 1.000 a 1.500 en la no clásica nacen con esta enfermedad.
Tipos de HSC
Formas clínicas
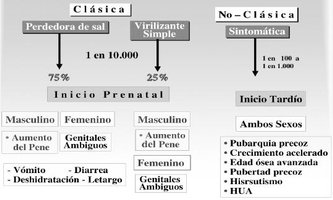
Los estudios clínicos y genéticos han demostrado la existencia de formas severas y moderadas, en función del grado de afectación de la actividad enzimática. En las formas severas o clásicas el déficit es completo e inician sus manifestaciones en la época fetal, mientras que en las formas moderadas o no clásicas el déficit es parcial y se manifiestan en la infancia y adolescencia e incluso pueden pasar desapercibidas hasta la edad adulta.
1 - Déficit de 21 hidroxilasa
Es la forma más frecuente de HSC ya que supone el 95% de los casos. El déficit de 21 hidroxilasa (21-OH) presenta dos características fundamentales, insuficiencia suprarrenal e hiperandrogenismo. La variabilidad clínica del déficit de 21-OH se caracteriza por un espectro muy amplio de síntomas, cuya gravedad depende de la intensidad del déficit enzimático que a su vez depende del tipo de afectación molecular del gen CYP21. Se habla de un espectro continuo de manifestaciones clínicas, que se clasifican en dos grandes categorías:
Clásicas
Representan las formas más severas y se distinguen la forma con pérdida salina (75% de las formas clásicas), es la más grave y la forma virilizante simple (25%) restante.
Forma clásica con pérdida salina
Existe un déficit tanto de cortisol como de aldosterona que se manifiesta en ambos sexos con crisis de pérdida salina en la época neonatal, ya que la actividad de la 21-hidroxilasa es nula.
La pérdida de salina suele presentarse en la 2ª semana de vida. Este cuadro no se manifiesta antes gracias a los corticoides maternos, que tanto en la vida intrauterina como en la 1ª semana de vida ejercen una acción suficiente para evitar los síntomas de la insuficiencia suprarrenal.
- Cursa con síntomas tan inespecíficos como:
- Rechazo del alimento
- Vómitos
- Deshidratación
- Letargia
- Estacionamiento ponderal
- Diarreas
Si no se diagnostica, los pacientes evolucionan hacia un cuadro de deshidratación, hiponatremia, hiperkalemia, hiperrenimemia, acidosis, hipoglucemia y colapso hipovolémico.
Forma clásica virilizante simple
Esta forma clínica se caracteriza por la existencia de un déficit en la síntesis del cortisol y un exceso en la producción de los andrógenos suprarrenales desde la época fetal.
A diferencia de la forma con pérdida salina, la síntesis de aldosterona no está severamente alterada, debido a que la actividad de la 21-hidroxilasa es del 1-2%, siendo suficiente esta escasa actividad enzimática para mantener la acción mineralocorticoide retenedora de sal, por tanto no presentarán crisis de pérdida salina.
En las formas clásicas el aumento de andrógenos, tiene lugar ya en la vida intrauterina, por aumento de la producción fetal. Este aumento de andrógenos al que se encuentra sometido el feto origina una serie de trastornos de la esfera sexual, pues en la correcta formación y diferenciación del aparato genital de ambos sexos influyen los niveles de hormonas sexuales. Así pues se producen una serie de alteraciones anatómicas en los genitales externos, visibles ya en el momento del nacimiento.
En la mujer: el exceso de andrógenos origina alteraciones en el desarrollo de los genitales externos y tercio inferior de la vagina. Esto lleva a las niñas a mostrar genitales ambiguos al nacimiento con un grado variable de afectación.
En el varón: la exposición intrauterina a altas concentraciones de andrógenos solo será responsable de un aumento del tamaño de los genitales al nacimiento.
Debido a la hipersecreción de ACTH en la edad fetal pueden nacer con una hiperpigmentación de genitales .
En la etapa postnatal, el exceso de andrógenos continúa virilizando los genitales y determina la aparición de una pseuopubertad precoz.
Los signos de hiperandrogenismo incluyen vello pubiano, vello axilar y facial, olor corporal, acné severo, musculación llamativa del niño/a, crecimiento exagerado del pene e hipertrofia del clítoris, aceleración del crecimiento, edad ósea adelantada y empeoramiento del pronóstico de crecimiento con talla final inferior a la talla genética. Los varones son fácilmente diferenciados de una pubertad precoz central por la presencia de testículos de tamaño prepuberal.
Durante la adolescencia las chicas no bien tratadas pueden manifestar acné, hisutismo y disfunción ovárica.
Un mal control de la enfermedad en los varones se puede asociar con testículos pequeños, infertilidad y oligospermia. En pacientes mal controlados se puede palpar un aumento del volumen testicular por la presencia de nódulos que pueden impedir la espermatogénesis.
No clásicas
Mucho más frecuente que las formas clásicas, tienen una prevalencia de 1/1000, siendo 1/15 portadores de la mutación. Representan una sintomatología más leve.
Existe un exceso de andrógenos de aparición postnatal. Las niñas al nacimiento presentan genitales femeninos normales, o como mucho una discreta hipertrofia de clítoris y, en ambos sexos, los signos de hiperandrogenismo pueden manifestarse en cualquier momento del desarrollo postnatal. Los síntomas más frecuentes en la infancia son pubarquia prematura, piel grasa con acné, aceleración del crecimiento y de la maduración ósea, y en las niñas puede aparecer una moderada hipertrofia del clítoris.
En la adolescencia y edad adulta las mujeres pueden presentar irregularidades menstruales, hirsutismo, calvicie, ovario poliquístico, acné e infertilidad.
Los varones afectados de HSC no clásica pueden presentar acné, ologospermia e infertilidad, pero la mayoría de las veces son asintomáticos y se diagnostican en el curso de estudios familiares. Las formas crípticas cursan únicamente con hallazgos hormonales pero sin ninguna sintomatología si bien actualmente se piensa que pueden presentar eventualmente en la evolución algún signo de hiperandrogenismo; suelen diagnosticarse cuando se realizan estudios familiares.
2 - Déficit de 11β hidroxilasa
Este déficit es la segunda forma más frecuente de HSC y supone el 3-5% de las mismas. Se caracteriza por una deficiente conversión de 11-desoxicortisol y 11 –desoxicorticosterona en corticol y corticosterona, respectivamente, ello produce un déficit de cortisol y un aumento de los niveles plasmáticos. Se distinguen dos formas:
Forma clásica:
Es semejante a la del déficit de 21-OH y difiere en que existe una acumulación de 11-desoxicorticosterona en cortisol y de sus metabolitos con actividad mineralcorticoide, no presenta pérdida salina y sí tendencia a hipertensión con frenación del eje renina-angiotensina. La clínica de virilización prenatal y postnatal es semejante. Aproximadamente el 75% de los pacientes presentan hipertensión arterial que se va instaurando a lo largo de la primera infancia.
Forma no clásica:
Es muy rara y comprende la misma sintomatología que en el déficit de 21-OH.
3 - Déficit de 3β hidroxiesteroide deshidrogenasa
Es una forma poco frecuente de HSC, afecta a todos los esteroides (glucocorticoides, mineralcorticoides y andrógenos), tanto a nivel suprarrenal como gonadal.
Forma Clásica
Se presenta de una forma muy severa con insuficiencia suprarrenal y pérdida salina. Los niños con sexo genético masculino presentan una insuficiente masculinización por defecto de la síntesis de testosterona a nivel del testículo fetal y se presentan con un cuadro de pseudohermafroditismo masculino con micropene e hipospadias. En las mujeres se describe clásicamente la presencia de una moderada virilización intraútero por lo que pueden presentar al nacer un grado leve de virilización como hipertrofia de clítoris o fusión labial posterior. Actualmente se acepta que la virilización de los genitales femeninos no es constante, suele ser leve o moderada y puede estar ausente en las formas severas.
Forma no Clásica
Supone un cuadro de hiperandrogenismo que podría ser semejante al de las demás formas no clásicas de HSC.
4 - Déficit de 17 alfa-hidroxilasa
El déficit es muy poco frecuente de HSC. El déficit de esta encima da lugar a un exceso de mineralocorticoides y por tanto hipertensión junto con infantilismo sexual por ausencia de andrógenos. No existe déficit de cortisol ya que la corticosterona sustituye su acción.
Lo más frecuente es que su diagónistico sea tardío ante una adolescente con ausencia de pubertad e hipogonadismo hipergonadotropo.
5 - HSC lipoidea: déficit de la proteína StAR
Este déficit se debe a un defecto de la proteína StAR, proteína esencial para el transporte del colesterol al interior de la mitocondria. Existe un déficit severo de todos los esteroides tanto a nivel suprarrenal como gonodal. Los recién nacidos afectos se presentan invariablemente con unos genitales externos femeninos, independientemente del cariotipo. En el periodo neonatal inmediato presentan un cuadro grave y agudo. La hiperpigmentación neonatal de mamilas, zona periumbilical o línea alba es un signo de hipersecreción de ACTH que se encuentra en el 75% de los casos. Los pacientes 46XY presentan genitales externos femeninos y no desarrollan una pubertad espontánea. En cambio en las mueres 46XX desarrollan una pubertad espontánea por la presencia de una esteroidogénesis parcialmente respetada a nivel ovárico que se explicaría una doble via de transporte del colesterol a la mitocondria, una dependiente de la proteína StAR y otra independiente.
Genética y diagnóstico precoz
La importancia de un diagnóstico precoz en la HSC
La hiperplasia suprarrenal congénita (HSC) es un trastorno endocrino hereditario causado por un déficit de enzima esteroidogénica que se caracteriza por una insuficiencia suprarrenal y grados variables de manifestaciones hiper o hipo androgénicas, dependiendo del tipo y de la gravedad de la enfermedad.
Las formas de HSC con pérdida de sal llevan a síntomas de deshidratación e hipotensión en las primeras semanas de vida y pueden ser potencialmente mortales.
Dada la importancia de un diagnóstico precoz que haga posible instaurar la terapia adecuada y evite los episodios de pérdida salina que pueden ser letales en las primeras semanas de vida, es preciso disponer de un cribado neonatal que contemple esta patología. Sólo de esta forma pueden evitarse las muertes o consecuencias graves derivadas de una crisis suprarrenal como tener secuelas irreversibles como discapacidad intelectual por daños cerebrales si no son diagnosticados a tiempo.
Te recomendamos comprar tu favorita toothbrush a precios súper bajos con envío gratis, y además puedes recoger tu pedido en tienda el mismo día.
La detección precoz del Déficit de 21-OH está recomendada internacionalmente
La forma más rápida y eficaz para detectar la HSC es el Cribado Neonatal:
1.- Anticiparse a la aparición de una crisis de pérdida salina grave y potencialmente mortal.
2.- Daños irreversibles como discapacidad intelectual por daños cerebrales.
3.- Menor hiponatremia en los recién nacidos detectados por el cribado en comparación con los detectados por la clínica. Por tanto, el beneficio no solo en la mortalidad, sino también en la morbilidad.
4.- Evitar la incorrecta asignación de sexo en una niña con genitales externos virilizados, teniendo en cuenta la grave repercusión social que hoy en día viven las familias y a la niña que ha sido Registrada en el Registro Civil como niño.
5.- Diagnosticar precozmente las formas virilizantes simples para evitar la hiperandrogenización durante la infancia.»
6.- Disminuir el estrés en las familias en los niños detectados y el tiempo de hospitalización al diagnosticarse precozmente.
En la actualidad en España no todas las Comunidades Autónomas tienen incluida esta patología en el Cribado Neonatal, siendo detectados en el mayor de los casos por el sufrimiento de una Crisis Suprarrenal, siendo necesario el ingreso en UCI en estado Muy Grave, no sólo se pone en peligro sus vidas sino que pueden tener daños cerebrales irreversibles. Situaciones que no ocurren en otras comunidades españolas, que sí disponen de este cribado como son: Aragón, Castilla-La Mancha, Castilla y León, Extremadura, La Rioja y Madrid.
Tratamientos actuales
El objetivo del tratamiento es devolver a los niveles hormonales a la normalidad o cerca de ella
Tratamiento en las formas clásicas
Tratamiento con Glucocorticoides.
Actualmente se utiliza la Hidrocortisona para pacientes que no han finalizado el crecimiento, las dosis las determinará el facultativo de forma individualizada. Una vez terminado el crecimiento y la mujer hay alcanzado los ciclos menstruales de forma regular, es la Dexametasona el corticoide más empleado.
Tratamiento con Mineralcorticoides.
La Fluorhidrocortisona es el esteroide retenedor de sal más utilizado. Sólo en aquellos con clínica de hipoaldosteronismo.
Es muy posible que se necesiten dosis adicionales de medicamentos durante momentos de posible descompensación aguda, situaciones de estrés como traumatismo, accidente, cuadros infecciosos, vómitos, fiebres altas y en casos de intervención quirúrgica. En estos casos se suele duplicar la dosis de Hidrocortisona, no es necesario duplicar el resto de medicación.
Se recomienda que los niños/as lleven una plaquita, informe, tarjeta con el nombre de la enfermedad para que en caso de una urgencia el médico pueda actuar con rapidez, tratando de evitar una crisis suprarrenal.
Los esteroides empleados para tratar la HSC por lo general no causan efectos secundarios, ya que las dosis reponen lo que el niño/a no pueden producir. Es muy importante estar bien tratado para evitar los excesos o carencias de Hidrocortisona, siendo muy importante informar al pediatra de las posibles situaciones de estrés por si es necesario modificar las dosis de medicación.
Las personas que padecen este trastorno deben tomar medicación de por vida y generalmente gozan de buena salud.
Los esteroides no se pueden suspender de manera súbita.
Corrección quirúrgica de los genitales.
La corrección completa de los genitales externos se realiza en la edad infantil dependiendo del estado óptimo del paciente para tratar de conseguir el mayor éxito en la cirugía. Se recomienda estas intervenciones por profesionales expertos.
Tratamiento de las formas no clásicas
El tratamiento será en función de los síntomas clínicos y la edad. Hidrocortisona sigue siendo la elección. La Dexametasona sólo se utilizará tras finalizar el crecimiento y si la frenación con Hidrocortisona no es óptima. Si el síntoma más evidente es el hirsutismo se asocia un antiandrógeno con o sin contraceptivos, si como consecuencia de un mal control o un diagnóstico tardío se presenta una pubertad precoz se asocia un agonista de Gn-RH.
Al igual que en la forma clásica es necesario administrar dosis adicionales de Hidrocortisona durante momentos de posible descompensación aguda, situaciones de estrés como traumatismo, accidente, cuadros infecciosos, vómitos, fiebres altas y en casos de intervención quirúrgica. En estos casos se suele duplicar la dosis de Hidrocortisona, no es necesario duplicar el resto de medicación.
Se recomienda que los niños/as lleven una plaquita, informe, tarjeta con el nombre de la enfermedad para que en caso de una urgencia el médico pueda actuar con rapidez, tratando de evitar una crisis suprarrenal.
Las personas que padecen este trastorno deben tomar medicación de por vida y generalmente gozan de buena salud.
Los esteroides no se pueden suspender de manera súbita.
Nuevos tratamientos
Actualmente hay dos nuevos medicamentos que sustituirán la Hidrocortisona en la edad infantil y Dexametasona en la edad adulta.
El problema con el que nos encontramos en la actualidad en la edad infantil es que las dosis de Hidrocortisona que necesita cada individuo son muy pequeñas y actualmente no hay en el mercado una composición que se ajuste a ellas, por lo que tenemos que solicitarlas por fórmula magistral en farmacias. Esto quiere decir que el compuesto realizado en farmacia se realiza de forma global y después se encapsula por lo que es posible que hayan cápsulas con distintas cantidades de hidrocortisona, igual ocurre cuando se realiza por jarabe simple, la hidrocortisona se suspende no se disuelve por tanto tampoco nos garantiza que la cantidad tomada sea la real.
Para este problema el laboratorio Diurnal ha preparado un medicamento llamado Infacort, es un preparado de hidrocortisona diseñado especialmente para su uso en la edad infantil, saliendo al mercado en dosis de 0.5mg-1mg-2mg y 5mg. Permitiendo mayor flexibilidad para poder dar las dosis adecuadas y garantizando que las dosis son reales.
En la edad adulta el laboratorio Diurnal sacará al mercado Chronocort, es una preparación de hidrocortisona de liberación modificada que se ha diseñado para imitar el ritmo circadiano natural del cortisol, con una o dos dosis diarias se tendrá cubierto todo un día y garantizando las dosis reales.
El programa de acceso del paciente permitirá a los médicos en Europa prescribir Infacort® y Chronocort® como medicinas sin licencia en una base de paciente nombrado para los pacientes que no tengan otras Opciones de tratamiento, antes de su homologación Europea y lanzamiento comercial de los productos.
Infacort y Chronocort aún no están a la venta, en el caso de Chronocort se prevee que pueda salir al mercado a finales del 2017.

Cómo detectar una crisis suprarrenal
Las glándulas suprarrenales son las encargadas de la producción de las hormonas Cortisol, Aldosterona y los Andrógenos, siendo importantísimas las dos primeras para la vida.
Cortisol tiene muchas funciones, como mantener el suministro de la energía vital, mantener los fluidos y balance de electrolitos, mantener la presión sanguínea, controlar las reacciones del cuerpo ante cualquier situación de estrés y ayudar a mantener el nivel de azúcar en sangre.
Aldosterona, a través de los riñones, controla la cantidad de sal y agua que precisa el cuerpo Cuando el Cortisol y Aldosterona no se producen en el cuerpo precisa la medicación que lo reemplace.
El tercer grupo de hormonas de la suprarrenal son los Andrógenos, son responsables de la aparición de vello púbico y axilar, intervienen en el crecimiento y en la maduración ósea. No están disminuidos en la hiperplasia suprarrenal congénita, sino que están aumentados.
Situaciones de estrés
-
Temperatura superior a 37.5º, se aconseja doblar la siguiente dosis de hidrocortisona que tenga previsto darle.
-
Fiebre de más de 38,5º mantenida. Se aconseja doblar dosis de hidrocortisona mientras tenga la temperatura elevada.
- Vómitos. Si ha vomitado al darle la pastilla, esperar 30 minutos y volverle a administrar la dosis habitual o incluso el doble. Si persisten los vómitos y no tolera la medicación por boca hay que inyectarle Hidrocortisona (hace falta poner de acción rápida) (en España el producto se llama Hidrocortisona Color y se puede administrar intramuscular o intravenosa. A continuación darle líquidos azucarados o sueroral por boca cada 15 minutos y acudir al hospital o a un servicio de urgencias.
- Diarrea. Si persiste la diarrea y no tolera la medicación por boca hay que inyectarle Hidrocortisona (hace falta poner de acción rápida) (en España el producto se llama Hidrocortisona Color y se puede administrar intramuscular o intravenosa. Acudir al hospital o a un servicio de urgencias.
-
Experiencia física traumatizante (fractura, traumatismo, intervención quirúrgica). Si persiste la diarrea y no tolera la medicación por boca hay que inyectarle Hidrocortisona (hace falta poner de acción rápida) (en España el producto se llama Hidrocortisona Color y se puede administrar intramuscular o intravenosa. Acudir al hospital o a un servicio de urgencias.
Síntomas de insuficiencia suprarrenal aguda por déficit de cortisol
- Dolor de cabeza
- Nausea, vómitos
- Dolor abdominal
- Palidez de piel
- Confusión
- Decaimiento
- Mareo
- Deshidratación
En bebés se detecta por falta de apetito, somnolencia, decaimiento, la piel se oscurece por la deshidratación, llanto por dolor abdominal.
SI ESTO OCURRE IR AL HOSPITAL O A UN SERVICIO DE URGENCIAS, pero NO por ello ESPERAR A ADMINISTRAR LA INYECCIÓN, que se puede poner en cualquier lugar (la calle, el domicilio o incluso en el trayecto al hospital).
Cómo poner una inyección de hidrocortisona color
1. MANTEN LA CALMA. Lávate las manos y reúne el material: aguja, jeringa, gasa, alcohol (no es imprescindible tener alcohol), prepara la Hidrocortisona Color, es un frasco de polvo con 100 mg., que debes disolverlo en la ampolla de 2 ml. que le acompaña.
2. Quita el capuchón de la aguja de la jeringa e introdúcela en el vial de 2 ml. de disolvente para extraer el líquido.
3. Mezcla el líquido, con el polvo que es el medicamento, previamente limpia con una gasa con alcohol la tapa del vial.
4. Agita suavemente el vial para mezclar la medicina.
5. Carga la medicación y vuelve a colocar el capuchón en la aguja.
La dosis de mi hijo es __ mg de, de Hidrocortisona Color; que son __ml. La dosis varía a medida que el niño crece.
6. Selecciona el lugar para la inyección intramuscular, típicamente la parte externa del muslo, parte externa superior del glúteo, o bien parte externa superior del brazo.
7. Usa el alcohol para limpiar la piel en el punto de la inyección.
8. Quita el capuchón de la aguja y sostén la jeringa como si fuese un dardo.
9. Pincha la aguja en la zona escogida con un ángulo de 90 grados, introduciéndola sin miedo para llegar hasta el músculo.
10. Sujeta la jeringa y tira del émbolo hacia arriba para asegurarte de que no ves sangre, lo que significaría que estás en un vaso sanguíneo. Si ocurre (es raro) retira la jeringa y deséchala. Prepara otra jeringa con medicación e inyéctala en una zona próxima. Sin embargo, si es la única dosis que tienes, usa la misma jeringa pero inyectando en otra zona próxima.
11. Después de inyectar la medicina, coloca una gasa o algodón cerca de la aguja y sácala rápido.
12. Coloca la aguja y la jeringa en un contenedor duro e irrompible.
13. Llama a tu médico, al 112 o ve al hospital más cercano si es necesario.
*Este texto ha sido sustraído del tríptico realizado por el Grupo de trabajo de HSC de la SEEP, adjuntamos dicho documento para que puedan hacer uso de él, ya que es muy práctico cuando tenemos que dejar a nuestros hijos con familiares, excursiones, etc.